
|
|
|---|
Chapter 1 - Basic Pharmacology
Definition of a Drug
an exogenous chemical not necessary for normal cellular function that significantly alters the function of certain cells when taken in fairly low doses.
Psychoactive Drugs
drugs that alter mood, thought, or behavior --most used to manage/treat psychopathology; some used recreationally.
Drug Naming Conventions
Chemical Name:
7-cloro-1,3-dihydro-1-methyl-5-phenyl-2H-benzodiazepin-2-one
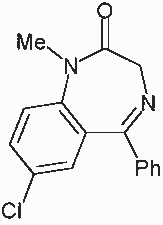
Generic Name (nonproprietary Name):
diazepam
Trade name (proprietary name):
Valium

Drug Doses: mg/kg (body weight)
Needed to standardize concentration of drug across subjects when measuring:
Drug Distribution parameters (Pharmacokinetics)
Drug Actions at target receptor proteins (Pharmacodynamics)
Behavioral changes induced by 1 and 2 above (Psychophysics)
Dose Response Curves (DRC)
A Cartesian Plot in which the X-axis plots concentration of a drug or hormone. The Y-axis plots response, which could be almost anything. For example, the response might be enzyme activity, accumulation of an intracellular second messenger, membrane potential, secretion of a hormone, heart rate or contraction of a muscle. Remember that the term "dose" strictly only applies to experiments performed with animals or people, where you administer various doses of drug. However, the term "dose-response curve" is also used more loosely to describe in vitro experiments where you apply known concentrations of drugs. Dose-response experiments typically use 10-20 doses of drug, approximately equally spaced on a logarithmic scale. For example doses might be 1, 3, 10, 30, 100, 300, 1000, 3000, and 10000 nM. When converted to logarithms, these values are equally spaced: 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0. Note: The logarithm of 3 is actually 0.4771, not 0.50. The antilog of 0.5 is 3.1623. So to make the doses truly equally spaced on a log scale, the concentrations ought to be 1.0, 3.1623, 10.0, 31.623 etc.

ED50 - median effective dose
LD50 - median lethal dose
TD50 - median toxic dose (clinical trials/human subjects)

DRCs and Drug Safety:
Therapeutic index (TI)= LD50 or TD50/ED50

Potency: differences on the ED50 of drugs that have the same effect
Effectiveness (efficacy): differences in the maximum effect of two drugs
Primary effects: The specific effect being measured at a specific point in time
Side effects: Other effects occuring concommitantly with a primary effect measurement
Drug Interactions
Antagonism: shifts the ED50 to the right
Additive effect: shifts ED50 to the left
Super additive effect (potentiation): Example would be Drug A having a completely flat DRC by itself-- but evidencing a dynamic dose response function in the presence of Drug B.
Pharmacokinetics
How drugs get into, around and out of the body.
Site of action: The point where you begin studying pharmacodynamics.
Routes of Administration: The first phase (step) in the study of pharmacokinetics
Parenteral (injection) Administration Routes
vehicle: Must be inert and compatable with the tissues being "invaded" by the needle.
s.c.: subcutaneous or "sub-q"-- the needle (and drug/vehicle bolus) is placed under the most superficial layers of skin but above the underlying muscle tissues-- results in slow, sustained pharmacokinetic absorbtion profiles.
i.m.: intramuscular -- the needle (and drug/vehicle bolus) is placed into the belly of large striated muscle tissues-- results in more rapid pharmacokinetic absorbtion profiles than sub-q.
i.p.: intraperitoneal -- the needle (and drug/vehicle bolus) is placed into peritoneal cavity containing all of the abdominal organs-- results in more rapid pharmacokinetic absorbtion profiles than sub-q or i.m.-- very rarely used in humans because of our unique abdominal musculature.
i.v.: intravenous --the needle (and drug/vehicle bolus) is placed directly into a vessel of the venous circulatory system-- results in extremely rapid pharmacokinetic profiles --in a sense absorbtion is "bypassed". In animal research a permantly inplanted i.v. catheter may be employed to examine behavioral phenomena such as drug self- administration (yes animals will use drugs recreationally and will even "pay" more for drugs they really "like").
diffusion:
substances tend to move from areas of high concentration to areas of low concentration until concentration is equal in both areas.
Inhalation:
Diffusion works between liquids and gasses --lungs are an efficient gas exchange system: oxygen in-CO2 out --Smoke and solids --snuff - tobacco, cocaine, heroin, etc.
Oral administration: (p.o.):
Digestive system --lipid barrier between food and blood; lipid solubility determines
absorption partition coefficient
Factors that determine lipid solubility
properties of the drug
ionization
Determinants of ionization:
1. pH of solvent
2. acidity/alkalinity of drug
3. pKa of drug
ION TRAPPING:
The tendency for acid drugs to get trapped on the basic side of a membrane and basic drugs to get trapped on the acid side of a membrane. Only nonionized molecules can diffuse through membrane. They reach equal concentration on both sides. The law of diffusion only applies to the nonionized molecules.Thus, only the concentration of nonionized molecules will be equal on both sides of a membrane.
This section is for those in class (and you know who you are) who needed to know "the mystical numbers" and how one obtains them -- you must not use this knowledge to harm your fellow earthlings or there will be retribution (I'll make you do the calculations on the exam without giving you the formulae --insert evil laughter here).
Acid Base Calculations and Solubility/Permeability of potential pharmaceutical compounds.
Acid/base chemistry is a very simple proton transfer equilibrium. A generic example where AH is any acid and A- is the conjugate base is given below.
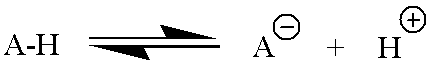
When we measure the pH of a solution, we are measuring the
concentration of hydrogen atoms (actually H3O+) in solution.
Strong acids will increase the number of hydrogen atoms and strong
bases will decrease the number of hydrogen atoms. By definition a
strong base is a weak acid (just look at the equilibrium above).
The equilibrium constant K is defined as the fraction of H+ and A- to the HA.
K = ([H+] [A-] ) / [HA] , for this equilibrium K is called Ka for acid dissociation constant.
Since we are interested in the amount of H+ we need to arrange the equation to just have [H+] in one side.
[H+] = Ka ([HA]/[A-] )
Taking the logarithm of each side gives:
log [H+] = log Ka + log ([HA]/[A-] )
Now we can multiple by -1.
-log [H+] = -log Ka - log ([HA]/[A-] )
Which is the same as:
-log [H+] = -log Ka + log ([A-] /[HA])
Traditionally, -log is substituted with a lower case p, so the equation becomes (known as the Henderson-Hasselback equation:
pH = pKa + log ([A-] /[HA])
So if we know the pH of a solution and the pKa the acid we can
determine how much [A-] and [HA] we have in solution, using the
rearranged equation below.
[A-] /[HA] = exp (pH - pKa)
This means that when pH is equal to pKa , [A-] = [HA], or that the acid is 50% ionized.
Example:
Drug, acid, pKa=3.5
Side A, pH=3.5,
ionization=50%, i.e, 1/2
Side B, pH= 7.5
ionization= .00001 i.e, 1/10,000
Now, only the nonionized molecules will be in equilibrium. This means that for each nonionized molecule on side A there will be only one ionized
molecule, but for each nonionized molecule on side B there will be 10,000 ionized molecules. Thus, there will be 10,000 more molecules on side B than side A.
Transdermal administration
epidermis packed with keratin
EG: nicotine patch
Distribution - where drugs go in the body.
Lipid solubility - highly lipid soluble drugs tend to concentrate in the lipids and water soluble drugs concentrate in body water.
Barriers:
Blood-Brain:
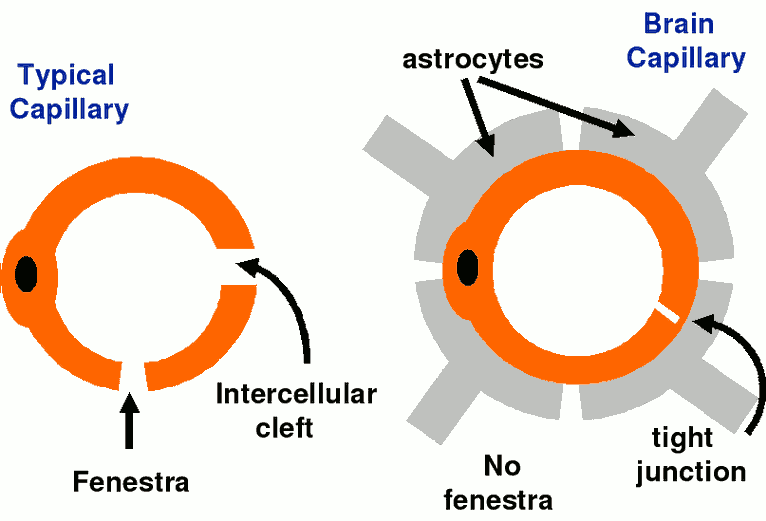
Placental:
protein binding:
Transport mechanisms:
active:
passive:
Excretion and metabolism:
Kidneys: filter everything out and reabsorb what is needed and what is lipid soluble. Reabsorption is altered by pH.
Liver: Chemical factory controlled by enzymes
enzymes:
- catalyst
- end in "ase"
Factors altering rate of metabolism:
1. Induction and blocking of enzymes
(antabuse blocks acetaldehyde dehydrogenase)
2. age
3. species
First pass metabolism: metabolism before drug is fully absorbed.
Combining absorption and excretion functions
Time course for drug concentration determined by rate of absorption and rate of excretion.
Different for different routes of administration. Half-life: time taken to reduce drug concentration by ½
Therapeutic window: Drug concentration above the therapeutic level and below the toxic level.
